図16 PP/CNF系をコアバックにより18倍発泡させた発泡体の断面27) ACS Applied Materials and Interfaces, 9(11), 9250-9254(2017)のFig. 2より引用
秋元英郎
秋元技術士事務所
化学発泡剤を用いた発泡成形は古くから行われていたが、物理発泡剤、特に超臨界流体を用いた発泡成形は過去20年で大きな進展を見せている。その20年のうち、前半の10年間は、学術目的にオートクレーブを用いたバッチ発泡による研究が多く行われ、産業的にはTrexel社のMuCell®技術(以降では微細発泡成形あるいは微細射出発泡成形と表示する)が市場投入され、称賛とともに迎えられた。ところが、実際にテストしてみると期待した効果が得られないといった落胆も生んだ10年であった。
その後の10年、すなわち直近の10年間は、微細射出発泡成形が自動車やプリンター等の情報家電分野でブレイクし、広く採用になるとともに、バッチではなく射出や押出の発泡プロセスを理解して知見を活用する目的の研究が多くなっている。
本稿では、過去10年間の発泡成形に関する研究や技術開発の成果を紹介する。
物理発泡剤を用いる発泡成形のプロセスにおいて、物理発泡剤をどのような状態でどの位置からどのようにして導入するかという課題は常に存在し、常に新しいプロセスが提案されている。
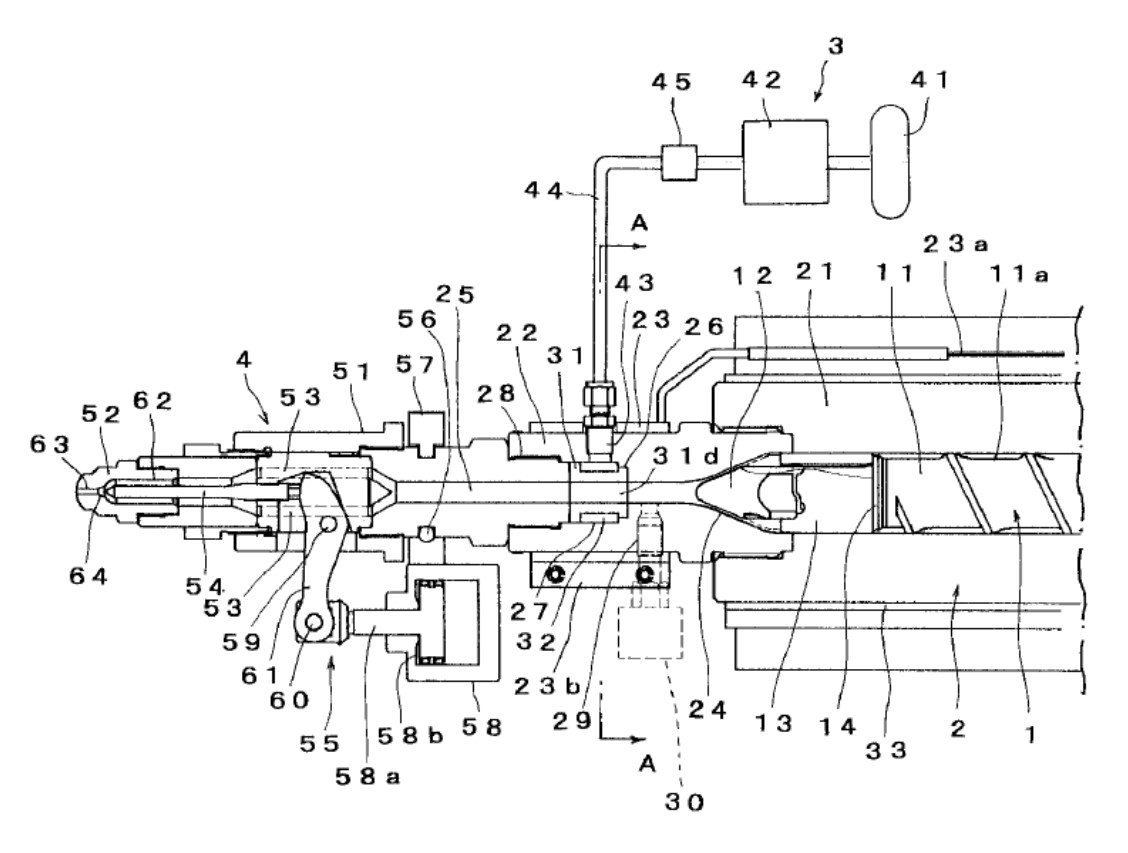
通常の物理発泡用射出成形機はスクリュー及びバレルを専用に設計する必要があるが、バレルとシャットオフノズルの間から発泡剤を導入する方法が提案されている1)。
図1において、物理発泡剤はバレルとシャットオフノズルの間に設けられた発泡剤導入装置の多孔質金属を通って溶融樹脂と接触し、金型内で発泡する。ミキシング工程を含まない点に特長を持つこの技術は、幕張メッセで開催されたIPF2017の東洋機械金属ブースで実演された。
一方で、可塑化ユニット全体を物理発泡剤で加圧し、可塑化の過程で溶融した樹脂に溶解させて発泡成形を行う取り組みもされている2,3)。この技術はK2016のArburgブース及びIKVブースで実演された。
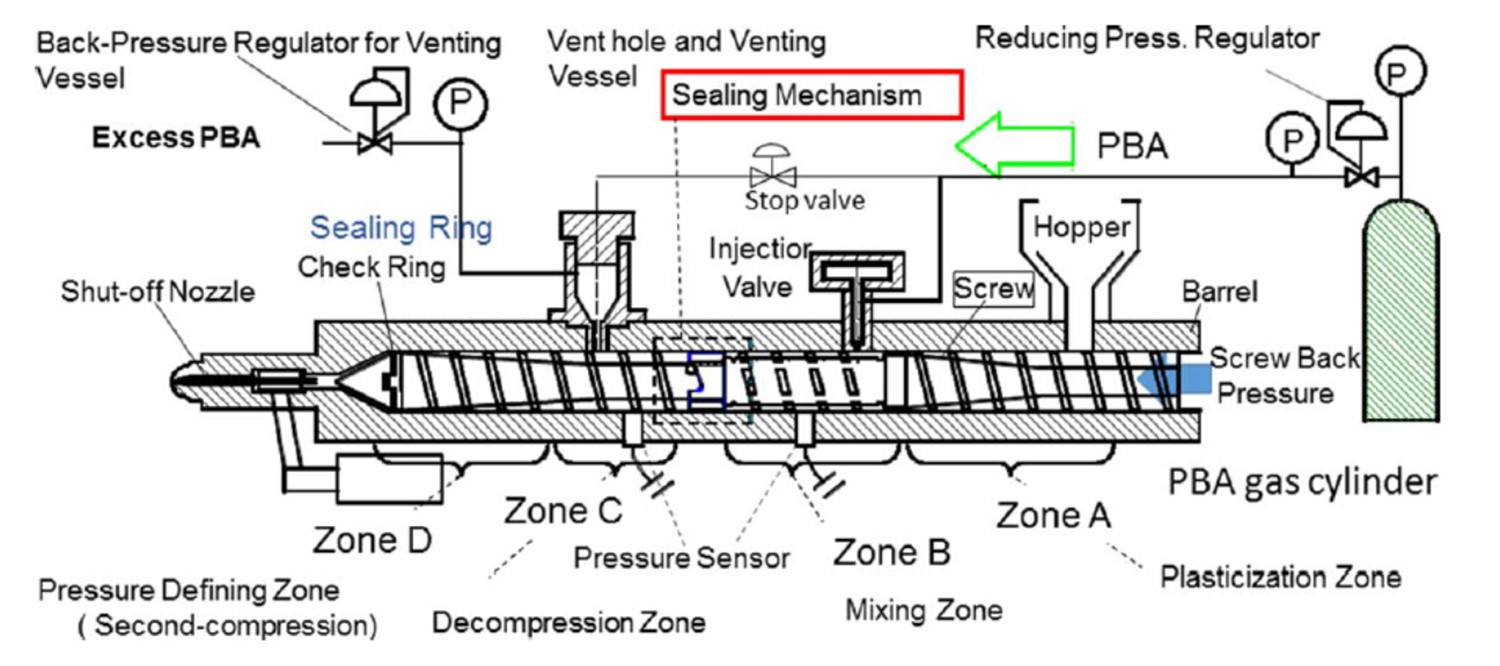
N2やCO2は超臨界状態で成形機に導入されるのが一般的であるが、昇圧ポンプ不要で物理発泡剤を成形機に導入するプロセスも提案されている。すなわち、図2に示すようにボンベから直接成形機のシリンダーに導入し、過剰な発泡剤をベントから排出して発泡剤の溶解量を制御する4)。
物理発泡剤として水を用いる方法も提案されている。通常の射出成形機に水の注入・混合部分を付け加えることで簡単に改造可能である。一例を示すと、所定の水を0.03秒という短時間に注入してミキシングノズルで混合する5)。気泡が発生する際の吸熱により成形品内部の除熱にも有利である。
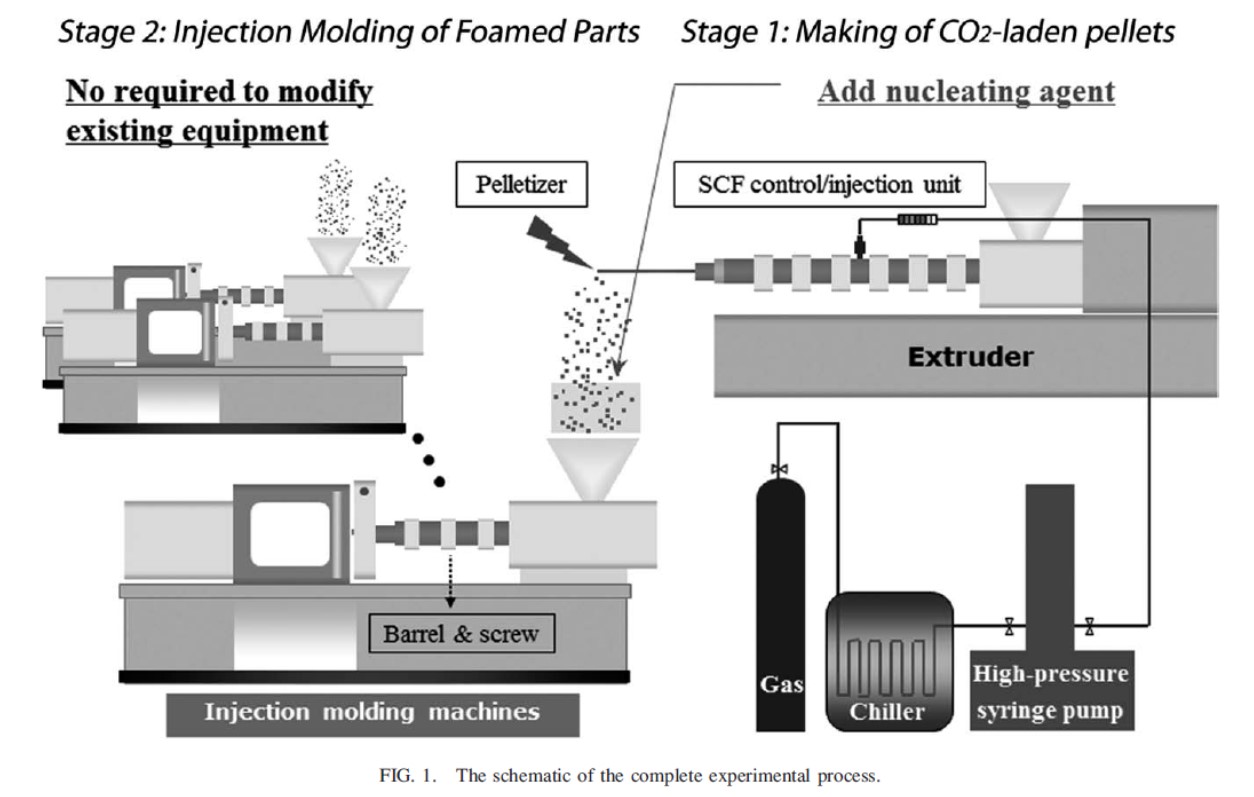
所定量のN2やCO2をペレットに含浸させておき、通常の成形機で成形することで発泡成形を行う試みも為されている。図3 に示すように、押出機を用いてN2やCO2を注入混合したペレットを用意すれば、射出成形機が何台あってもそれらは改造不要である6)。
図3 発泡剤含浸ペレット製造と発泡剤含浸ペレットを用いた発泡成形6)
Polymer Engineering and Science, 51(11), 2295-2303(2011)のFig.1より引用
N2約0.2%、CO2約0.5%含浸したペレットを放置すると、造粒後の時間経過とともに徐々に発泡性を失うが、図4に示すようにCO2はN2に比べてシェルフタイム(大気中でペレットが発泡性を維持する時間)が長い7)。おそらく、CO2の方がN2よりもマトリックス樹脂との親和性が高いからであろう。
物理発泡成形において、発泡体の形状(ソリッドスキン層厚み、平均気泡径、気泡密度、気泡径分布、気泡形状)は成形条件に大きく影響される。
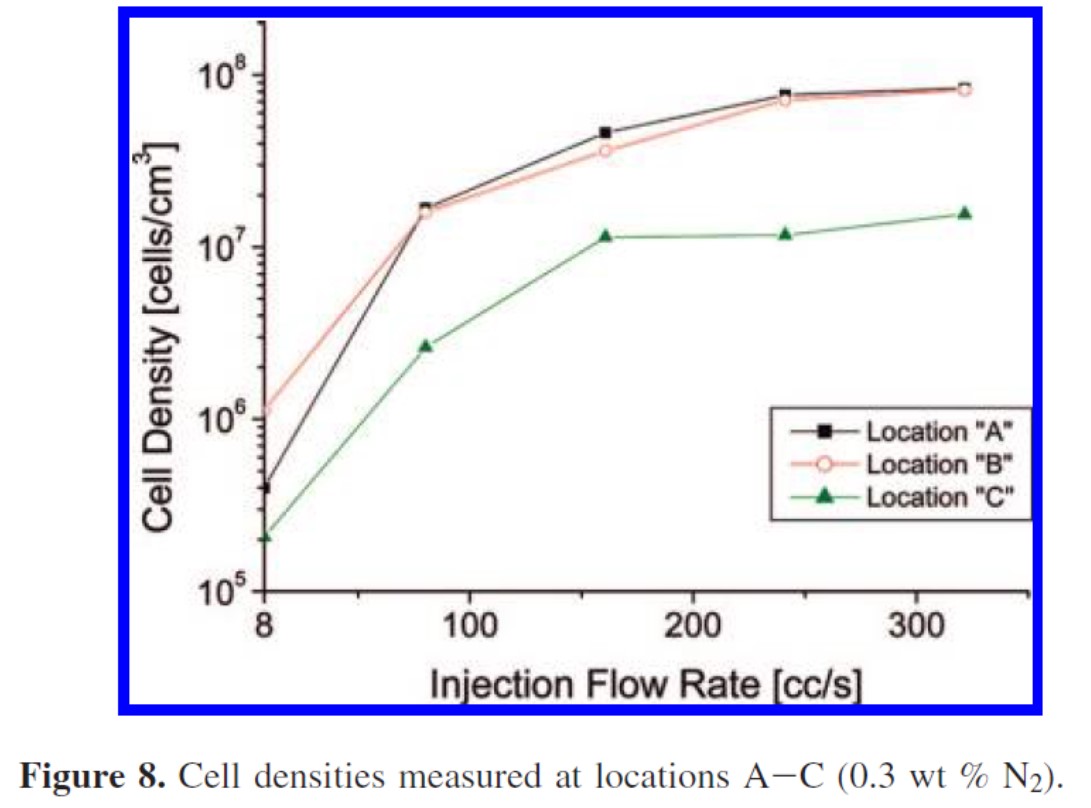
同じショットサイズ(計量値)で比較すると射出率が大きいほど気泡密度が高くなる(図5)とともに金型充填率が高まる。逆に射出率が小さいとショートショットになりやすい。発泡剤量が多いほどゲート通過時の圧力降下が大きく、気泡核が生成しやすい。
図5 射出率と気泡密度の関係 A:ゲート付近,B:中央部,C:末端部8) Industrial and Engineering Chemistry Research, 47(23), 9460(2008)のFig. 8より引用
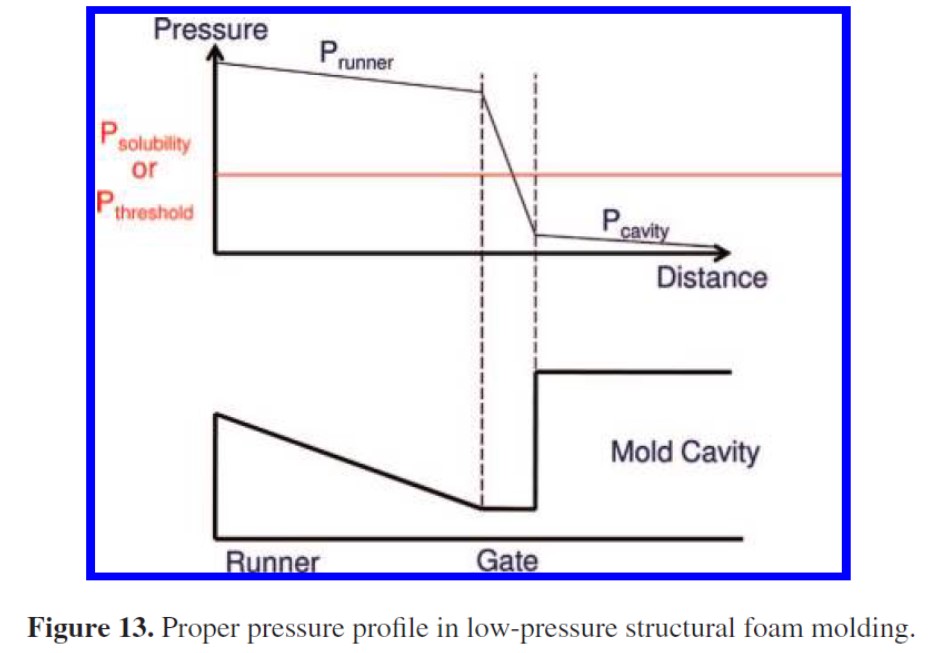
また、発泡倍率を高めるには図6に示すようにゲートにおける抵抗(圧力損失)を大きくする必要がある8)。図中では、ゲート通過時に樹脂圧力(黒線)が飽和圧力(赤線)を横切り、過飽和になる様子を示している。
図6 ゲート形状(図下)とゲート前後における圧力パターン(図上)
(図の例ではゲート通過時に飽和圧力より下がっている)8) Industrial and Engineering Chemistry Research, 47(23), 9463(2008)のFig. 13より引用
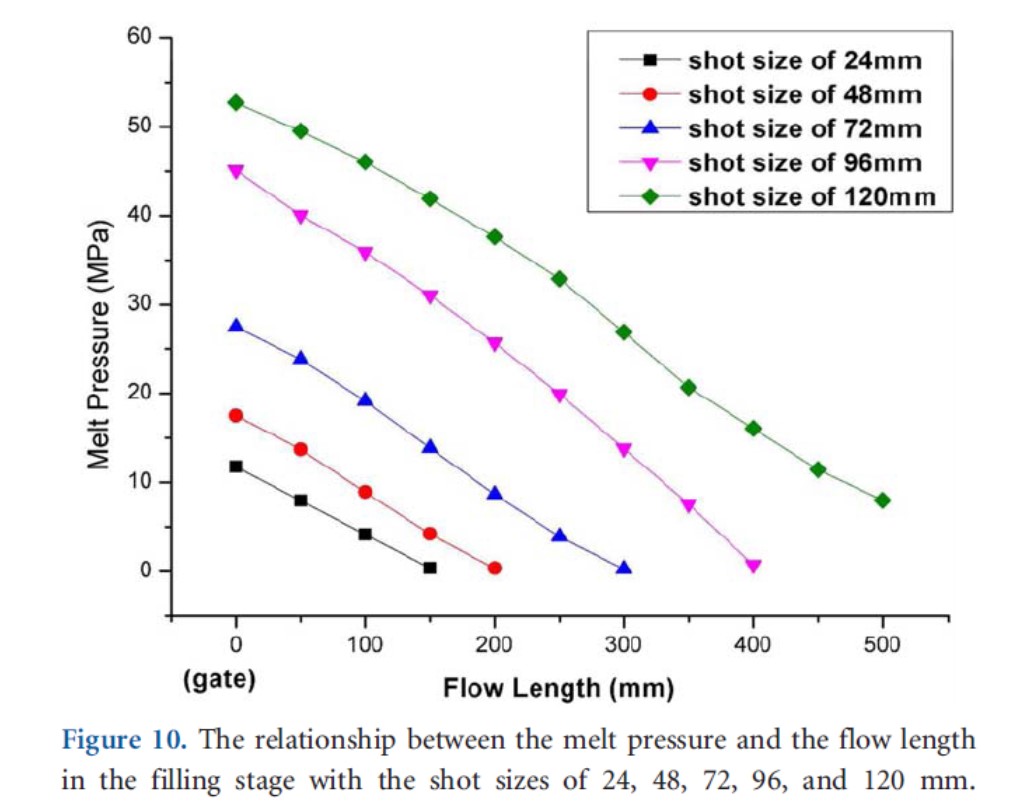
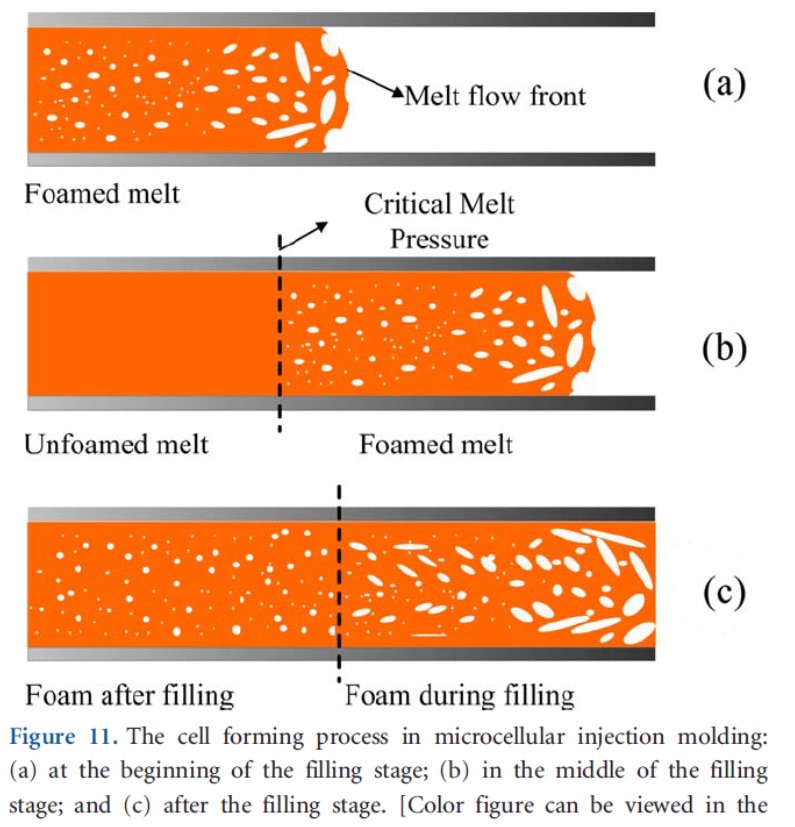
充填時における金型内圧力が発泡剤の飽和圧力を超えるか超えないかは成形品の気泡構造に大きな影響を与える。ABS樹脂の微細射出発泡成形において計量値を振って成形を行うと、図7に示すようにゲートから離れるほど金型内圧力は下がり、計量値が大きいほど同じ位置での圧力は高くなる。計量値がある値以上になると図8に示すように金型内圧力が飽和圧力より高くなり、充填過程で気泡が発生せず、充填完了後に気泡が発生する部分ができる9)。
図8 金型内を流れる溶融樹脂と気泡の様子9) (a):流動初期,(b):流動途中であり、ゲートに近い部分で飽和圧力を超える
(c):充填完了後であり、ゲートに近い部分も飽和圧力より下がる
Journal of Applied Polymer Science, 131(12), 40365(2014)のFig. 11より引用
図9にはHIPS(耐衝撃性ポリスチレン)を用い、金型内圧力を5MPaで成形した場合と30MPaで成形した場合の成形品の断面写真を示す。原文献では考察していないが、5MPaではゲートで生成した気泡がそのまま残り、30MPaで見られるのは充填完了後に発生た気泡であろう10)。
ガスカウンタープレッシャー(GCP)とコアバックを組み合わせて、PCの高倍率・高連続気泡率の発泡体が得られている11)。樹脂温度、射出率、コアバック量が気泡形状に大きく影響し、発泡倍率8倍、連続気泡率85%の発泡体が得られた。
気泡壁をフィブリル化することによる連続気泡化についても研究されている。ゲル化剤やポリプロピレンの結晶核剤として知られている1,3:2,4 ビス-O-(4-メチルベンジリデン)-D-ソルビトールをPPにブレンドし、N2を発泡剤として用いてコアバック法で高倍率の発泡体にすると、気泡壁で結晶化が進んでナノスケールにフィブリル化して、連通率が90%以上の連続発泡体が得られる12)。
また、結晶化促進効果を持つソルビトール系化合物を用いずに、コアバック遅延時間を調整してPPの結晶化温度付近でコアバックを行うことでも連通率が高い発泡体が得られる13)。
連続気泡の高倍率発泡体の想定される用途一つとして吸音材が考えられる。PCの連続発泡体でソリッド材の2.5倍の吸音特性が得られたとの報告もある14)。
射出発泡成形では、金型内で気泡の発生,成長,消失,合一,破裂が起こっている。成形条件によって実際に何が起こっているかを可視化によって観察することは、発泡成形を理解するためには極めで有効である。
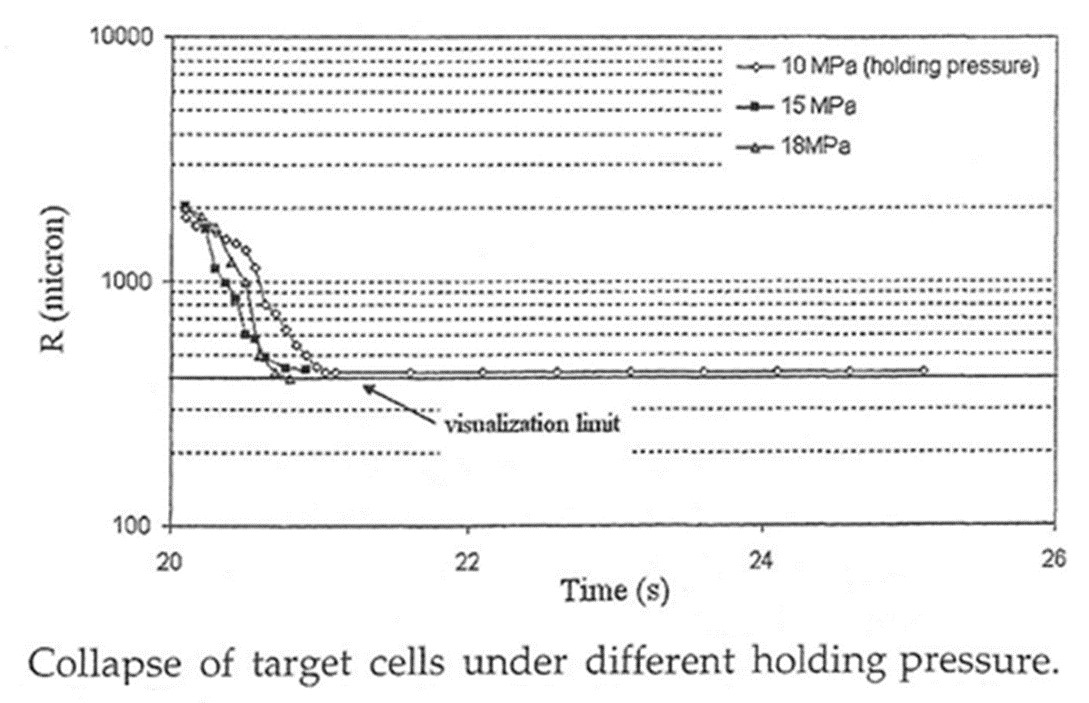
GPPSに0.5%のCO2を発泡剤として使い、気泡の成長と消滅の様子を可視化実験によって観察した例を示す15)。気泡は400μmが観察限界である。充填初期では発生した気泡は樹脂の流動よりも速く流れて末端に達する傾向にある。
キャビティが樹脂で充填されるとキャビティ内圧力が高まり、気泡は消滅する。このとき、圧力勾配により流動下流に気泡が残る傾向にある。図10には射出充填後の冷却過程で発生する気泡の径の時間変化を示した。図11 は気泡が成長し終わった後に保圧を掛けて気泡径の変化・消失を観察したものであり、圧力により気泡の消失が起こることが理解できる。
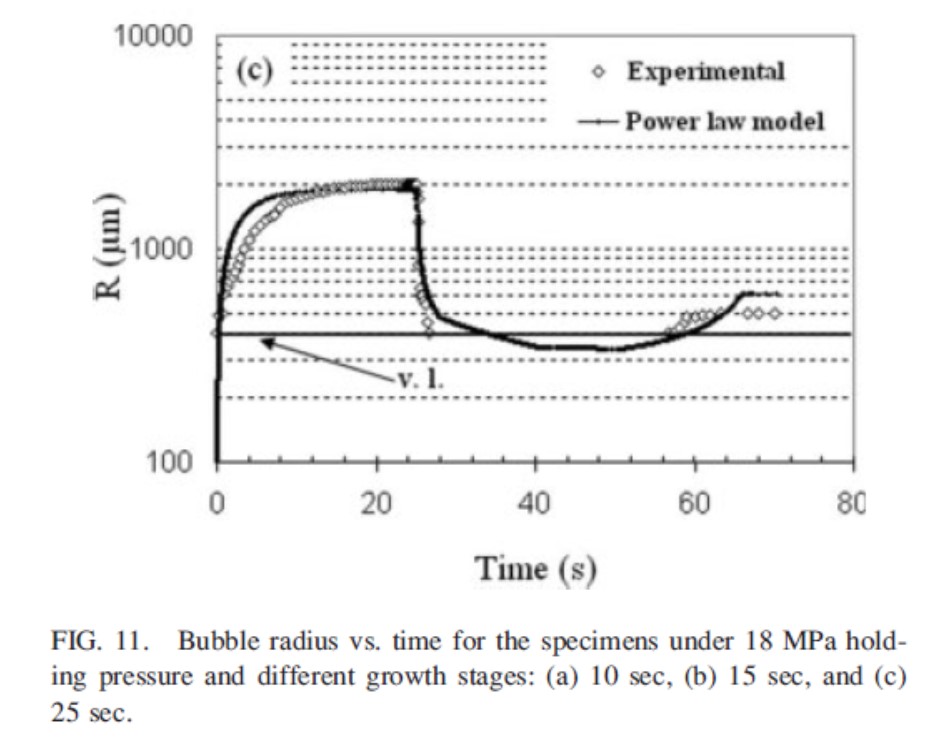
しかしながら、気泡の消失はあくまで可視化の観察限界よりも気泡径が小さくなったことで見えなくなっただけであり、気泡が消滅したわけではないと考えられる。保圧過程で消失した気泡がその後の冷却過程で再度出現することが観察されている。図12には射出充填から冷却までの時間に対する気泡径の変化の実測値とPower Lawモデルによる計算値を示した。観察限界よりも小さい気泡が消滅せずに残っていたと考えられる16)。
図12 射出から冷却までの気泡径の変化の実測値と計算値16)
v.l.は可視化実験による気泡検出限界
Polymer Engineering and Science, 50(3), 561-569(2010)のFig. 11より引用
射出発泡成形において気泡核生成に2つのメカニズムが存在することが可視化実験で確認されている。1つはゲート通過の際の圧力降下によるものであり、もう1つは充填後の冷却による収縮過程における圧力低下によるものである。ゲートで生成した気泡は充填が進むに従い金型内圧力が高まることで消失するが、ゲートからの距離が遠くなると圧力が伝わりにくく、ゲートで生成した気泡が消失せずに残ることがある17)。
図13にはゲート近傍と製品中央部における気泡の様子の比較を示した。ゲートから遠い位置では、消失せずに残った気泡と冷却過程で生成する気泡が混在することがわかる。
図13 流動長の中央部(上)とゲート付近(下)における気泡の様子の経時変化17) 上の矢印はゲートで生成して残った気泡
European Polymer Journal, 76, 2-13(2016)のFig. 3の(b)とFig. 5より引用
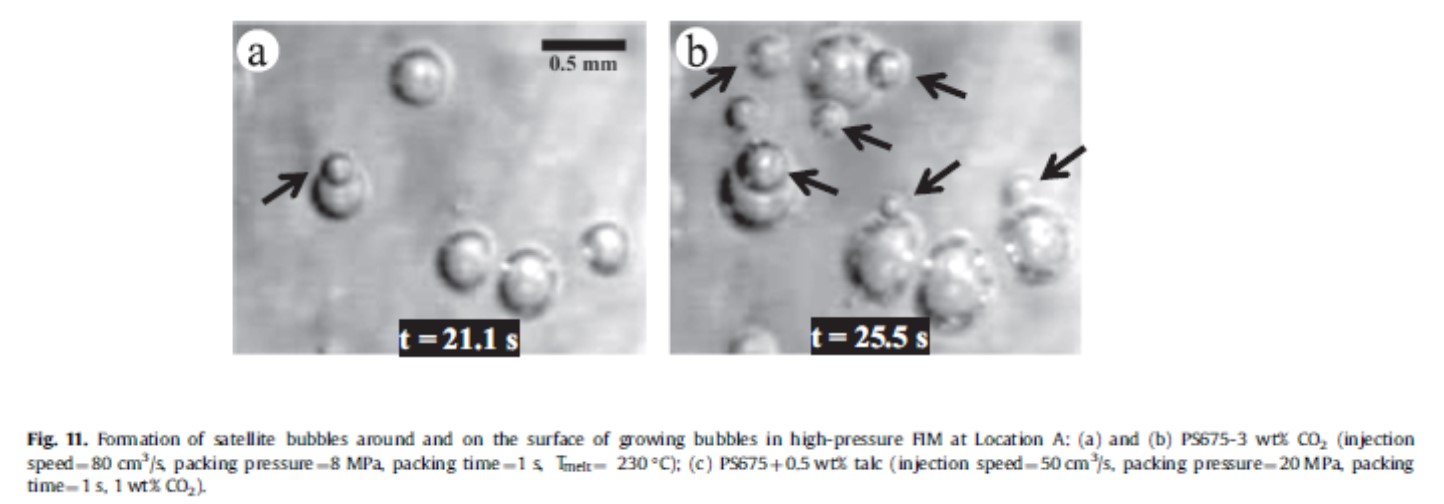
生成した気泡の周辺は気泡壁に位置する分子が配向することで応力が残留している可能性があり、気泡の周辺から新たな気泡が発生する様子も観察されている(図14)18)。
図14 既に存在している気泡の周辺に新たに気泡が発生する様子の写真18)
矢印で新たに発生した気泡を示す
Chemical Engineering Science, 155, 27-37(2016)のFig. 11より引用
発泡成形が難しいとされるスーパーエンプラの発泡成形にも取り組まれている。PEEKはそのままでは発泡成形することが難しいが、カーボンナノファイバーを添加すると良好な発泡体が得られることが報告されている19)。
PEIに多層型カーボンナノチューブ(MWCNT),ナノクレー,タルクなどのフィラーを添加して成形したところ、MWCNTを添加したものでPEIより気泡径が小さく、気泡密度が大きくなった20)。
また、PEIにPPをブレンドすると、未変性PP添加の場合に気泡径が30μmであったのに対し、無水マレイン酸変性PP添加の場合に気泡径は10μmになった21)。
熱膨張型マイクロカプセル型の発泡剤は古くから存在したが、発泡成形に用いる場合には、成形温度とマイクロカプセルが膨張する温度を合わせる必要がある。
マイクロカプセルは炭化水素をポリアクリロニトリル等のガスバリア性樹脂シェルで包んでいる。カプセルの膨張特性はシェル材の粘弾性特性に依存し、粘弾性特性を最適化することにより200℃で成形するPPに使用できるマイクロカプセルが得られた22)。
シェル材の最適特性を得るにはガス透過性と耐破裂性、膨張性を最適化できるシェル材の架橋度、tanδがあり、最適な架橋度を達成するためにアクリロニトリルに不飽和カルボン酸を共重合し、亜鉛等の金属イオンによる架橋を行っている23)。
タルクは発泡核剤としての効果を持つフィラーとして知られている。PPの発泡成形におけるタルクの核剤効果の研究として、粒子径,粒子径分布,粒子形状,表面処理の違いが気泡核生成にどのように影響するかが検討されている。粒子径や表面積によって発泡体の気泡密度が2倍以上の違いとなって現れる24)。
ナノサイズに分散したクレー(ナノクレー)にも核剤効果が見つかっている。PPに無水マレイン酸変性PPを相溶化剤としてナノクレーのコンポジットを作成し、発泡成形を行ったところ、ナノクレーの添加量が2%で、劇的な核剤効果が得られた25)。
セルロースナノクリスタル(NCC)やセルロースナノファイバー(CNF)の効果に関する研究も盛んである。PPにNCCを添加すると、気泡径が小さく気泡密度が大きくなった26)。その一例を図15に示す。
図15 PPの発泡に対するナノセルロースクリスタルの効果26) (a):NCC無添加,(b):NCC 3wt%添加
Journal of Applied Polymer Science, 132(47), 42485(2015)のFig. 2より引用
CNFをPPにブレンドするとコアバック法で高倍率に発泡させることが可能になる。最大で18倍発泡が得られ(図16)、セルロースの繊維は気泡が延びる方向に向いた構造になる27)。
図16 PP/CNF系をコアバックにより18倍発泡させた発泡体の断面27)
ACS Applied Materials and Interfaces, 9(11), 9250-9254(2017)のFig. 2より引用
CNFはPPの結晶化にも影響を与えることが分かってきた。無水アルケニルコハク酸で処理したCNFを1~5wt%添加したPPをコアバックで高発泡させると、空隙率が最大で80%の発泡体が得られた。CNFの添加によりPPの結晶化温度が上昇するとともに、発泡体の気泡径が減少し、気泡密度が増加した28)。
繊維状のフィラーを樹脂にブレンドして成形するとフィラーは流動方向に配向する傾向があり、導電性フィラーを用いた場合には導電性に異方性が生じる。その一方で、導電性フィラーをブレンドした樹脂を発泡成形すると違った挙動が見られる。
PSに炭素繊維(CF)をブレンドしてCO2を用いて発泡させると、図17に見られるように気泡の拡大の影響を受けて、平行移動と回転運動を行って繊維の向きが変わる29)。
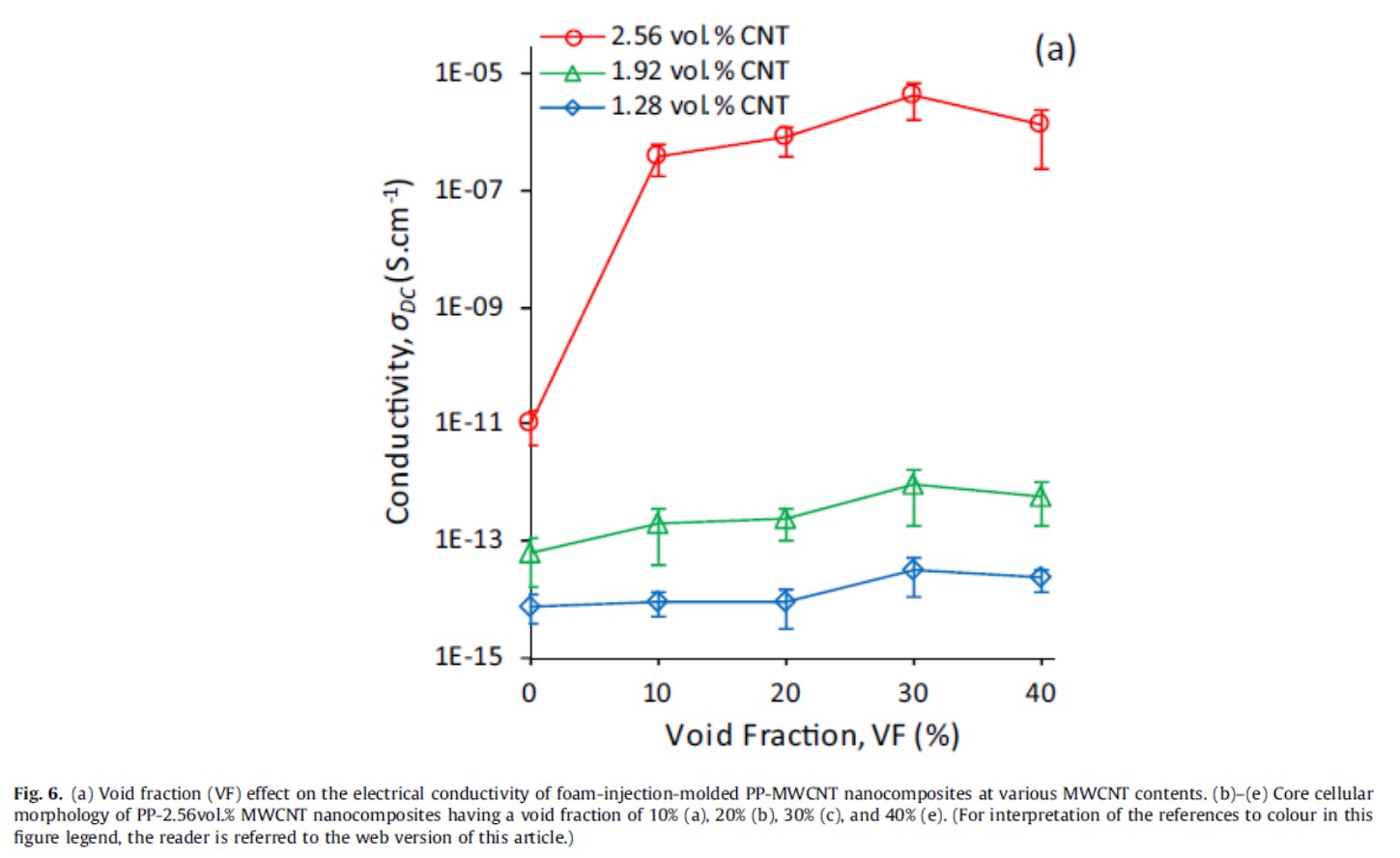
PPにMWCNTを2.56 vol%添加したコンポジットの電気伝導率をソリッドと発泡品で比較すると、ソリッドに比べて空隙率10%品で約5桁、空隙率30%品で約6桁の電気伝導率の向上が見られた(図18)30)。
図18 異なる量のカーボンナノチューブをブレンドした発泡PPの導電性30)
Composites Part A: Applied Science and Manufacturing, 96, 28-36(2017)のFig. 6 (a)より引用
微細射出発泡成形では成形品表面にスワールマークと呼ばれる特有の外観不良が現れることが知られている。スワールマークを生じさせないためにはヒート&クール成形と組み合わせると効果があることはよく知られているが、金型温度をどの程度まで上げればよいかの指標は十分ではなかった。
成形品の表面粗さに着目すると、表面粗さが改善する温度領域があることがわかる。図19の例では、電磁誘導による金型加熱を行っているが、100℃までは改善効果が無く、100℃~160℃の間では金型温度ともに表面粗さが改善し、180℃以上に上げてもそれ以上の改善は見られない31)。
図19 電磁誘導加熱を用いたヒート&クール成形における金型温度と表面粗度の関係31)
Advances in Polymer Technology, 27(4), 224-232(2008)のFig. 4より引用
発泡成形(特に超臨界流体を用いた発泡成形)は直近の10年で急速に採用事例が拡大してきている。本稿では紙面の都合上触れなかったが、発泡専用材料の開発も盛んになっている。シンプルな設備の開発により、設備投資が軽減される可能性もあり、今後の用途拡大が大いに期待される分野である。
1)特開2012-232558
2)Michaeli W., Krumpholz T., Obeloer D.,Tech. Papers, Region. Tech. Conf. – Soc. Plast. Eng.,2,1019-1023(2008)
3)Michaeli W., Hopmann C., Obeloer D.,Conf. Proc. Soc. Plast. Eng.,2,1551-1556(2011)
4)Yusa A., Yamamoto S., Goto H., Uezono H., Asaoka F., Wang L., Ando M., Ishihara S., Ohshima M., Polym. Eng. Sci., 57(1), 105-113(2017)
5)Escudero J., Solórzano E., Rodriguez-Perez M.A., Garcia-Moreno F., De Saja J.A.,Cell. Polym.,28(4), 289-302(2009)
6)Lee J., Turng L.-S., Dougherty E., Gorton P., Polym. Eng. Sci., 51(11), 2295-2303(2011)
7)Sun X., Turng L.-S. Polym. Eng. Sci.,54(4), 899-913 (2014)
8)Lee J.W.S., Wang J., Yoon J.D., Park C.B., Ind. and Eng. Chem. Res., 47(23), 9457-9464 (2008)
9)Dong G., Zhao G., Guan Y., Wang G., Wang X., J. Appl. Polym. Sci., 131(12), 40365(2014)
10)山田岳大,村田泰彦,横井秀俊,成形加工,21(10), 633-639(2009)
11)Jahani D., Ameli A., Cofreros A., Park C.B., Naguib H.E., Conf. Proc. Soc. Plast. Eng., 2, 1692-1696(2013)
12)Miyamoto R., Yasuhara S., Ohshima M.,Soc. Plast. Eng. – 2013 SPE Int. Polyolefins Conf. (2013)
13)Chu R.K.M., Mark L.H., Jahani D., Park C.B., Conf. Proc. Soc. Plast. Eng., 3, 2544-2548(2014)
14) Jahani D., Ameli A., Saniei M., Ding W., Park C.B., Naguib H.E., Macromol. Mater. Eng., 300(1), 48-56(2015)
15) Mahmoodi M., Behravesh A.H., Rezavand S.A.M., Pashaei A., J. Appl. Polym. Sci., 116(6), 3346-3355(2010)
16)Mahmoodi M., Behravesh A.H., Rezavand S.A.M., Golzar M., Polym. Eng. Sci., 50(3), 561-569(2010)
17) Shaayegan V., Wang G., Park C.B., Eur. Polym. J., 76, 2-13(2016)
18) Shaayegan V., Wang G., Park C.B., Chem. Eng. Sci., 155, 27-37(2016)
19) Verdejo R., Werner P., Sandler J., Altstädt V., Shaffer M.S.P., J. Mater. Sci., 44(6), 1427-1434(2009)
20) Li J., Chen Z., Wang X., Liu T., Zhou Y., Luo S., J. Appl. Polym. Sci., 130(6), 4171-4181(2013)
21)Liu T., Chen Z., Lei Y., Wang X., Luo S., J. Cell. Plast., 51(4), 387-400(2015)
22)Kawaguchi Y., Ito D., Kosaka Y., Nakachi T., Kake H., Kim J.K., Shikuma H., Ohshima M., Conf. Proc. Soc. Plast. Eng., 2, 914-919(2009)
23)Kawaguchi Y., Ito D., Kosaka Y., Okudo M., Nakachi T., Kake H., Kim J.K., Shikuma H., Ohshima M., Polym. Eng. Sci., 50(4), 835-842(2010)
24)Meli G., Jouffret F., Dave P., Conf. Proc. Soc. Plast. Eng., 1, 216-223(2014)
25)Rizvi S.J.A., Bhatnagar N., Int. Polym. Proc., 26(4), 375-382(2011)
26)Yousefian H., Rodrigue D., J. Appl. Polym. Sci., 132(47), 42485(2015)
27)Wang L., Ishihara S., Hikima Y., Ohshima M., Sekiguchi T., Sato A., Yano H., ACS Appl. Mater. Interfaces, 9(11), 9250-9254(2017)
28)Wang L., Ando M., Kubota M., Ishihara S., Hikima Y., Ohshima M., Sekiguchi T., Sato A., Yano H., Composites Part A: Appl. Sci. Manuf., 98, 166-173(2017)
29)Shaayegan V., Ameli A., Wang S., Park C.B., Composites Part A: Appl. Sci. Manuf., 88, 67-74(2016)
30)Ameli A., Kazemi Y., Wang S., Park C.B., Pötschke P., Composites Part A: Appl. Sci. Manuf., 96, 28-36(2017)
31)Chen S.-C., Lin Y.-W., Chien R.-D., Li H.-M., Adv. Polym. Tech., 27(4), 224-232(2008)
This website uses cookies.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}